CHAPTER 8
Thermal Expansion
As indicated earlier, addition of thermal motion displaces the inter-atomic equilibrium in the outward direction. A direct effect of the motion is thus an expansion of the solid structure. This direct and positive result is particularly interesting in view of the fact that previous theories have always been rather vague as to why such an expansion occurs. These theories visualize the thermal motion of a solid as an oscillation around an equilibrium position, but they fail to shed much light on the question as to why that equilibrium position should be displaced as the temperature rises. A typical “explanation” taken from a physics text says, “Since the average amplitude of vibration of the molecules increases with temperature, it seems reasonable that the average distance between the atoms should increase with temperature.” But it is not at all obvious why this should be “reasonable.” As a general proposition, an increase in the amplitude of a vibration does not, in itself, change the position of equilibrium.
Many discussions of the subject purport to supply an explanation by stating that the thermal motion is an anharmonic vibration. But this is not an explanation; it is merely a restatement of the problem. What is needed is a reason why the addition of thermal energy produces such an unusual result. This is what the Reciprocal System of theory supplies. According to this theory, the thermal motion is not an oscillation around a fixed average position; it is a simple harmonic motion in which the inward component is coincident with the progression of the natural reference system, and therefore has no physical effect. The outward component is physically effective, and displaces the atomic equilibrium in the outward direction.
From the theoretical standpoint, thermal expansion is a relatively unexplored area of physical science. Measurement of the expansion of different substances at various temperatures is being pursued vigorously, and the volume of empirical data in this field is increasing quite rapidly. However, the practical effect of the change in the coefficient of expansion due to temperature variation is of little consequence, and for most purposes it can be disregarded. As stated in the physics text from which the “explanation” of the expansion was taken, “Accurate measurements do show a slight variation of the coefficient of expansion with the temperature. We shall ignore such variations.” This lack of significant practical application has limited the amount of theoretical attention that the subject has heretofore received. But one of the principal objectives of this present work is to demonstrate that the Reciprocal System is a general physical theory. However limited the practical use of the thermal expansion information may be, we will want to show that this expansion can be explained on the same basis as the other properties of matter, using the same principles and relations that are applied to those other properties, with only such modifications as are required by considerations peculiar to the expansion.
In general, the volumetric behavior of a solid in response to the application of heat is analogous to that of a confined gas, the differences being limited to those items which depend on whether the point of equilibrium between any two of the constituent atoms is inside or outside unit distance. At constant pressure, the general gas equation (5-3), which describes the relation between the principal properties of the ideal gas, reduces to:
| V = kT |
(8-1) |
|
This is Charles’ Law. It tells us that at constant pressure the volume of an ideal gas (one that is entirely free from time region forces) is directly proportional to the absolute temperature.
The relation E = PV (equation 4-3) is merely a restatement of the definition of pressure, in a different form, and is therefore valid in the time region (inside unit distance) as well as in the ideal gas state. Since E = kT2 (equation 5-5) in the time region, it follows that in this region:
| PV = kT2 |
(8-2) |
|
At constant pressure this reduces to:
| V = kT2 |
(8-3) |
|
In our consideration of volume changes in solid structures due to the addition of thermal energy we will usually be interested mainly in the coefficient of thermal expansion, or derivative of volume with respect to temperature. This is obtained by differentiating equation 8-3.
| dv/dT = 2kT |
(8-4) |
|
Aside from the numerical constant k, this equation is identical with the specific heat equation 5-7, where the value of n in that equation is unity. Thus there is a close association between thermal expansion and specific heat up to the first transition temperature defined in Chapter 5. For all of the elements on which sufficient data are available to enable locating the transition point, this transition temperature is the same for thermal expansion as it is for specific heat. Each element has a negative initial level of the expansion coefficient, the magnitude of which has the same relation to the magnitude at the transition point as in specific heat; that is, 2/9 in most cases, and 1/9 in some of the electronegative elements. It follows that if the coefficient of expansion at the transition point is equated to 4.63 specific heat, the first segment of the expansion curve is identical with the first segment of the specific heat curve.
Beyond the transition point the thermal expansion curve follows a course quite different from that of the specific heat, because of the difference in the nature of the two phenomena. Since the term n3 is absent from the thermal expansion equation, the modification of the expansion curve that takes place where motion of single units is succeeded by multi-unit motion involves a change in the coefficient k. The expansion is related to the effective energy (that is, to the temperature), irrespective of the relation between total energy and effective energy that determines the specific heat above the first transition point. The magnitude of the constant K that determines the slope of the upper segment of the expansion curve is determined primarily by the temperature of the end point of the solid state.
For purposes of this present discussion, the solid end point will be regarded as coincident with the melting point. As brought out in Chapter 7, this is, in fact, only an approximate coincidence. But the present examination of thermal expansion is limited to its general features. Evaluation of the exact quantitative relations will not be feasible until a more intensive study of the situation is undertaken, and even then it will be difficult to verify the theoretical results by comparison with empirical data because of the large uncertainties in the experimental values. Even the most reliable measurements of thermal expansion are subject to an estimated uncertainty of ±3 percent, and the best available values for some elements are said to be good only to within ±20 percent. However, most of the measurements on the more common elements are sufficiently accurate for present purposes, as all that we are here undertaking to do is to show that the empirically determined expansions agree, in general, with the theoretical pattern.
The total expansion from zero temperature to the solid end point is a fixed quantity, the magnitude of which is determined by the limitation of the solid state thermal motion (vibration) to the region within unit distance. At zero temperature the gravitational motion (outward in the time region) is in equilibrium with the inward progression of the natural reference system. The resulting volume is s03, the initial molecular volume. At the solid end point the thermal motion is also in equilibrium with the inward progression of the natural reference system, as this is the point at which the thermal motion is able to cross the time region boundary without assistance from gravitation. The thermal motion up to the end point of the solid state thus adds a volume equal to the initial volume of the molecule. Because of the dimensional situation, however, only a fraction of the added volume is effective in the region in which it is measured; that is, outside unit space.
For an understanding of the dimensional relations that are involved it is necessary to realize that all of the phenomena of the solid state take place inside unit space (distance), in what we have called the time region. The properties of motion in this region were discussed in detail at appropriate points in Volume I. This discussion will not be repeated here, but a brief review of the general situation, with particular reference to the dimensions of motion may be helpful. According to the fundamental postulates of the Reciprocal System, space exists only in association with time as motion, and motion exists only in discrete units From this it follows that space and time likewise exist only in discrete units. Consequently, any two atoms that are separated by one unit of space cannot move any closer together in space, as this would require the existence of fractional units. These atoms may, however, accomplish the equivalent of moving closer together in space by moving outward in time. All motion in the time region, the region inside unit space, is motion of this kind: motion in time (equivalent space) rather than motion in actual space.
The first unit of thermal motion is a one-dimensional motion in time. At the transition point, T1, this motion has reached the full one-unit level. As already explained, only half of this unit is physically effective. One fully effective unit is required for escape from the time region, and the motion therefore enters a second time region unit. In this second unit a three-dimensional distribution of the motion is possible. But the motion in time that takes place in the time region has only a scalar connection with motion in the region outside unit space, which is motion in space. This is equivalent to a one-dimensional contact. Thus only one dimension of the three-dimensional time region motion is effective beyond the regional boundary. The effective fraction of the motion is 1/8 of one unit, or 1/16 of the total two-unit time region motion. The expansion is proportional to the effective component of the motion, and this means that the volumetric expansion from zero temperature to the solid end point, as measured in the region outside unit space, is also 1/16, or 0.0625 of the initial volume. On a one-dimensional (linear) basis, this is 0.0205.
This is the relative expansion that would take place providing that no change in the volumetric determinants of the substance occurs above the reference temperature (usually room temperature). But such changes occur more often than not, and, as has been explained, the volume changes accompanying an increase in temperature are normally in the direction of increased volume. The total expansion is 0.0625 of the initial volume corresponding to the volume at the solid end point. Where this theoretical initial volume is greater than the reference volume projected to zero temperature, the expansion expressed relative to the smaller volume is correspondingly increased. It follows that in most cases the linear expansion, as measured, is somewhat above 0.0205, generally in the range from this value up to about 0.028.
The increase in volume at the higher temperature, where it occurs, is generally due to structural rearrangements. The changes take place either in the inter-atomic distance, by reason of transitions from one of the types of orientation discussed in Chapter 1 to another, or in the crystal structure, or both. The expansion is related to the inter-atomic distance (s0) rather than to the geometrical volume, and it is independent of the geometrical arrangement, but, as indicated in the preceding paragraph, a modification of the geometry does affect the relation of the volume at the solid end point to the reference volume at zero temperature.
In the NaCl type of structure the edge of the unit cube is equal to the inter-atomic distance. This cube contains one atom, and the ratio of the measured volume to what we may call the three-dimensional space, the cube of the inter-atomic distance, is therefore unity. In the body-centered cube the edge is 2√3 times the inter-atomic distance. Since the unit cube of this type contains two atoms, the ratio of volume to three-dimensional space is 0.770. The one-dimensional space, the edge of a hypothetical cube containing one atom, is then 0.9165 for the body-centered cube and 1.00 for the NaCl type structure. Transitions from one type of structure to the other modify the spatial relations accordingly. The values applicable to all five of the principal isometric crystal structures of the elements are listed in the following tabulation.
| Face-centered cube | 0.8909 | |
| Close-packed hexagonal | 0.8909 | |
| Body-centered cube | 0.9165 | |
| Simple (NaCl) cube | 1.0000 | |
| Diamond (ZnS) cube | 1.1547 | |
The second segment of the thermal expansion curve has no negative initial level, because there is a positive expansion (that of the first segment) into which the initial level can extend. Like the transition from the liquid to the solid state, the transition from single units of motion to multi-unit motion involves a change in the zero datum applicable to temperature. The temperature T0, corresponding to the initial negative level, is eliminated, and the temperature of the end point, T1, of the first segment of the curve, which is 9/2 T0 on this segment, is reduced to 7/2 T0 on the second segment.
As brought out in Chapter 7, the minimum of the zero point temperature, T0, is equivalent to one of the 128 dimensional units that correspond to one full temperature unit, 510.8 degrees K. As the temperature rises, additional units of motion are activated, and the corresponding value when all 128 units are fully effective is thus 7/2 × 510.8 = 1788 degrees K. Under the same maximum conditions, the second unit of thermal motion, from T1 to the solid end point, adds an equal magnitude. Thus the temperature of this theoretical full-scale solid end point is 3576 degrees k. The total expansion coefficient at T1 on the first segment of the expansion curve, and at the initial point of the second segment, is then 0.0205/3576. However, this coefficient is subject to a 1/9 initial level. This makes the net effective coefficient 8/9 × 0.0205/3576 = 5.2×10-6 per degree K.
Where the end point temperature (which we are equating to the melting point, Tm, for present purposes) is below 3576, the average coefficient of expansion is increased by the ratio 3576/Tm, inasmuch as the total expansion up to the solid end point is a fixed magnitude. If the first temperature unit, up to T1, were to take its full share of the expansion, the coefficient at T1on the first segment of the expansion curve, and at the initial point of the second segment, would also be increased by the same ratio. But in the first unit range of temperature the thermal motion takes place in one time region dimension only, and there is no opportunity to increase the total expansion by extension into additional dimensions in the manner that is possible when a second unit of motion is involved. (Additional dimensions do not increase the effective magnitude of one unit, as 1n = 1.) The total expansion corresponding to the first unit of motion (speed) can be increased by extension to additional rotational speed displacements, but this is possible only in full units, and is limited to a total of four, the maximum magnetic displacement.
As an example, let us consider the element zirconium, which has a melting point of 2125° K. The melting point ratio is 3576/2125 = 1.68. Inasmuch as this is less than two full units, the expansion coefficient of zirconium remains at one unit (5.2×10-6) at the initial point of the second segment of the curve, and the difference has to be made up by an increase in the rate of expansion between this initial point and Tm; that is, by an increase in the slope of the second section of the expansion curve. The expansion pattern of zirconium is shown graphically in Figure 14.
Figure 14: Thermal Expansion
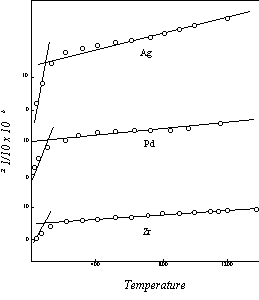
Now let us look at an element with a lower melting point. Titanium has a melting point of 1941° K. The ratio 3576/1941 is 1.84. This, again, is less than two full units. Titanium therefore has the same one unit expansion coefficient at the initial level as the elements with higher melting points. The melting point of palladium is only a little more than 100 degrees below that of titanium, but this difference is just enough to put this element into the two unit range. The ratio computed from the observed melting point, 1825° K, is 1.96, and is thus slightly under the two unit level. But in this case the difference between the melting point and the end point of the solid state, which we are disregarding for general application, becomes important. as it is enough to raise the 1.96 ratio above 2.00. The expansion coefficient of palladium at the initial point of the second segment of the curve is therefore two units (10.3×10-6), and the expansion follows the pattern illustrated in the second curve in Fig.14.
The effect of the difference between the solid end point and the melting point can also be seen at the three unit level, as the melting point ratio of silver, 3576/1234 = 2.90, is raised enough by this difference to put it over 3.00. Silver then has the three unit (15.5×10-6) expansion coefficient at the upper initial point, as shown in the upper curve in Fig.14. At the next unit level the element magnesium, with a ratio of 3.87, is similarly near the 4.00 mark, but in this instance the end point increment is not sufficient to close the gap, and magnesium stays on the three unit basis.
None of the elements for which sufficient data were available for comparison with the theoretical curves has a melting point in the four unit range from 715 to 894° K. But since the magnetic rotation is limited to four units, the four unit initial level also applies to the elements with melting points below 715° K. This is illustrated in Figure 15 by the curve for lead, melting point 601° K.
Figure 15: Thermal Expansion
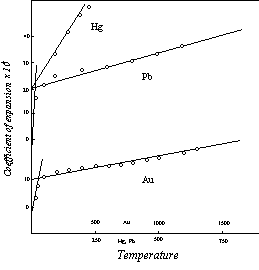
As can be seen in Figure 14, the expansion coefficient of silver, as measured experimentally, deviates from the straight line relation in the vicinity of T1. This deviation is not due to experimental error or to structural readjustments. It is a result of the nature of the transition from the one unit expansion below T1 to the multi-unit expansion above this temperature. Unlike the specific heat transition, where the increments represented by the second segment of the specific heat curve add to the specific heat at T1, the expansion represented by the second segment of the expansion curve replaces the expansion represented by the first segment. The initial level of the second segment at zero temperature is the unit (or n-unit) level reached at the end of the first segment.
This means that at T1 the molecule undergoes an isothermal expansion to the level of the second segment at that temperature. In the aggregate the individual molecular expansions are spread out over a temperature range by the distribution of molecular velocities, and they appear as a bulge in the expansion curve. Coincidentally, there is a downward deviation in the curve, similar to that in the experimental specific heat curves, due to the effect of the transition to the more nearly horizontal second segment of the curve. The net effect of these two types of deviation from the theoretical curve applying to the single molecule depends on their relative magnitude, and on the temperature range over which the deviations are distributed. The curves of Figure 14 have been selected from among those in which the net deviation is at a minimum, in order to minimize uncertainties in the definition of the upper sections of the curves, and to make it clear that these linear sections actually terminate at the calculated initial levels. More commonly, the bulge is quite pronounced, as in the curves for gold and lead, Figure 15.
When the effect of this systematic deviation from the .linear relation in the vicinity of the transition point is taken into consideration, all of the electropositive elements included in the compilation of expansion data utilized in the investigation,12 except the rare earth elements, have expansion curves that follow the theoretical pattern within the range of accuracy of the experimental results. Most of the rare earths have the one unit expansion coefficient (5.2×10-6) at the initial level of the second segment of the curve, although their melting points are in the range where coefficients of two, or in some cases three, units would be normal. The reason for this, the only deviation from the general pattern in the expansion curves of these elements, is as yet unknown, but it is no doubt connected with the other peculiarities of the rare earth elements that were noted earlier.
The electronegative elements of Division III follow the regular pattern. The lowest melting point in this group is that of mercury, 234° K, well below the lowest value for any of the electropositive elements investigated, but this descent to a lower melting point does not introduce any new behavior. The upper segment of the expansion curve for mercury, defined by the empirical data in Fig.15, definitely terminates at the four unit level (20.7×10-6), as required by the theory. Thus the theoretical relations are applicable to the full temperature range of the first three divisions.
As noted earlier, the borderline elements of Division IV, those with negative electric displacement 4, are capable of acting as members of either Division III or Division IV. The expansion curve for lead, Fig.15, follows the normal Division III pattern. The lower borderline elements, tin and germanium, have curves in which the initial levels, like those of the rare earths, are lower than the values corresponding to the melting points. Otherwise, these curves are also normal. Very little is known about the expansion of the elements of negative displacement below 4. The theoretical development has not yet been extended to a consideration of the effect of the strongly electronegative character of these elements on the volume relations, and the empirical data are both meager and conflicting.
This Division IV situation is part of the general problem of anisotropic expansion, a subject to which the Reciprocal System of theory has not yet been applied. The measurements previously cited that apply to anisotropic crystals were made on polycrystalline material in which the expansion in different directions is averaged as a result of the random orientation in the aggregate. Both this issue of anisotropic expansion and the application of the thermal expansion theory to compounds and alloys are still on the waiting list for future investigation. There is no reason to believe that such an investigation will encounter any serious difficulties, but for the present other matters are being given the priority.